- Joined
- Apr 26, 2010
- Messages
- 218
- Reaction score
- 0
In the lab where I do research, I do enzyme assays. In a nutshell, I am testing various inhibitors on proteins. We are testing inhibitors for a protein necessary for a parasite that causes Chaga's disease to live.
So my part is to test it on the human enzyme and see which one inhibits it the least, while my partner is testing it on the parasite enzyme, looking for one that inhibits it the most.
The problem is that we are both getting inconsistent results. My controls, despite me being very very careful, are not consistent. It may have to do with the activity of the enzyme (since it is kept in the freezer, I take it out and thaw it in ice. Perhaps I have to thaw it longer even though it is clearly thawed out?).
We are trying to find the Ki (forgot what it stands for) of the inhibitors using a Lineweaver Burk plot. But the thing is my PI is getting frustrated (when I plotted my points they didn't align well linearly). The reaction rate is supposed to decrease linearly as more inhibitor is added. He said to me that if I can't figure out what is going on, he's gonna give up on trying to find the Ki and do something simpler. But the thing was he told me before he was planning on publishing the Ki data 🙁
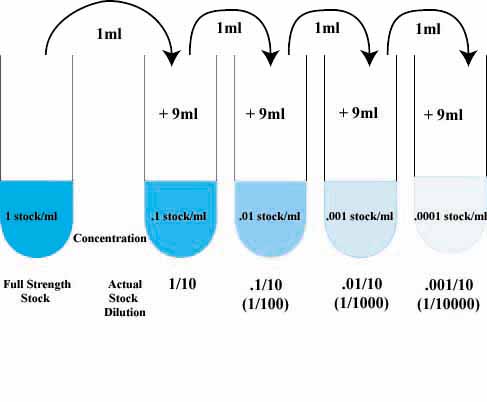
So what should I do? I'm being careful as it is. Since we use very small amounts of protein (~1 microliter), we tried diluting it five fold and then using five microliters of the dilution to minimize error from extra protein sticking on the pipette tip, but still not consistent.
I'm not sure if I put this in the right forum, but can anyone with experience doing enzyme assays and troubleshooting help me out here? I've been working on this since June and I really would like to get this Ki done. Thanks!
So my part is to test it on the human enzyme and see which one inhibits it the least, while my partner is testing it on the parasite enzyme, looking for one that inhibits it the most.
The problem is that we are both getting inconsistent results. My controls, despite me being very very careful, are not consistent. It may have to do with the activity of the enzyme (since it is kept in the freezer, I take it out and thaw it in ice. Perhaps I have to thaw it longer even though it is clearly thawed out?).
We are trying to find the Ki (forgot what it stands for) of the inhibitors using a Lineweaver Burk plot. But the thing is my PI is getting frustrated (when I plotted my points they didn't align well linearly). The reaction rate is supposed to decrease linearly as more inhibitor is added. He said to me that if I can't figure out what is going on, he's gonna give up on trying to find the Ki and do something simpler. But the thing was he told me before he was planning on publishing the Ki data 🙁
So what should I do? I'm being careful as it is. Since we use very small amounts of protein (~1 microliter), we tried diluting it five fold and then using five microliters of the dilution to minimize error from extra protein sticking on the pipette tip, but still not consistent.
I'm not sure if I put this in the right forum, but can anyone with experience doing enzyme assays and troubleshooting help me out here? I've been working on this since June and I really would like to get this Ki done. Thanks!